
Nowadays, there is almost no active medical device without some interaction with software. Since the introduction of the European Medical Device Regulation 2017/745 (MDR) and In-Vitro Diagnostic Medical Devices Regulation 2017/746 (IVDR), the industry talks about the impact and classification of Medical Device Software.
Medical Device Software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a "Medical Device" in the MDR or IVDR.
More precisely, if software is used in the context of an illness, injury or disability for the purpose of monitoring, prediction, diagnosis, alleviation, prognosis, therapy, the control of conception or in-vitro diagnostics. Medical device software can be a standalone device (i.e. Mobile Medical App) as well as software which is part of a medical device. If software is intended to drive or influence the use of a (hardware) medical device and does not have or perform a medical purpose of its own, nor create information on its own, it is considered to be embedded software of a Medical Device
While the above definition seems to be clear, it must be understood that Medical Device Software has to be differentiated between Medical Device Software embedded in Medical Devices and Software as a Medical Device (SaMD). Importantly, the definition of Software as a Medical Device, does not refer to the related platform and/or technology (i.e. mobile app, cloud, server, etc.).
Software as a Medical Device is intended to be used for one or more medical purposes and performing these purposes without being part of a medical device (non-embedded). In other words, a standalone software product.
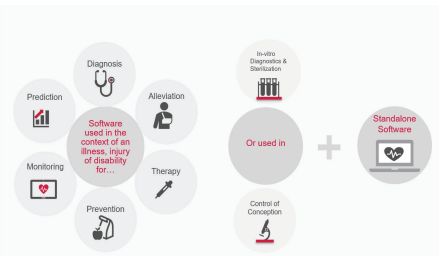
Figure 1: Illustration of Medical Device Software & Software as Medical Device
1 International - Regulatory Requirements
In an international context it is very important to think about all the country-specific regulatory requirements that may be applicable wherever you intend to market the software.
Depending on the country where you intend to submit your Medical Device Software application you need to consider the local requirements with respect to the design, development and operation of the software. Most countries require the international standard IEC 62304 (Medical device software – software life cycle processes). This standard is recognized by the EU, Australia, Canada and in the US. China has implemented an equivalent standard YY/T-0664 (Medical Device Software) as a recommendation for Medical Device manufacturers. It is important to wisely choose an accredited test laboratory (either a National Certification Body or an accredited Testing Laboratory) for the purpose of certification to IEC standards. Depending on the company's regulatory strategy and the corresponding countries where it is intended to market the Medical Device Software product, consideration should be given to requesting a CB scheme format which includes the testing of the country-specific deviations at an accredited test laboratory. The CB scheme format supports the mutual recognition of testing reports from accredited test laboratories in different countries.
The recently published standard IEC 82304 (Health Software) is not yet binding in order to market a device but can be considered by your Notified Body when it comes to "state of the art" discussions. Companies should be prepared to justify why this standard has been ignored. Basically, this standard is only applicable for Standalone Medical Device Software. It should also be noted that this standard is not only applicable for "Medical" devices. As suggested by the title, this standard is to be applied to "Health" software, meaning any kind of apps which inform you about health status (i.e. fitness trackers, body weight, nutrition apps). IEC 82304 refers to the development lifecycle of IEC 62304. Differences focus mainly on the accompanying documentation of Health Software products and the change control of such products in the post-market phase.
In Europe the classification of a Medical Device product needs to be considered in accordance with MDR, rule 11. For many Medical Device Software manufacturers, the introduction of MDR necessitates upgrading their existing software classification (under Medical Device Directive 93/42/EEC) to a higher risk class. Manufacturers who want to enter the Medical Device Software market, would be advised to consider a proper strategy, helpful in fulfilling the requirements of MDR. Even software with both a medical and a non-medical intended purpose need to cumulatively comply with the requirements applicable to devices with an intended medical purpose and those applicable to devices without an intended medical purpose (see MDR, Article 1, subparagraph 3).

Click here to continue reading . . .
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
