

Çeşitli bireysel hassasiyetler ve inançlar sebebiyle bağışıklama (immunisation) çalışmalarında istenen seviyede yaygın uygulanamasa da aşılama faaliyetleri, günümüzde hastalıkların önlenmesi bakımından birincil yöntem olarak görülmekte ve kamu sağlığı önlemleri bakımından en düşük maliyetle en güçlü korumayı sağlamaktadır . En basit şekliyle aşılar, bağışıklık sistemini uyararak hastalığa karşı koruma sağlayan biyolojik ürünler olarak tanımlanmaktadır . Aşılamadaki temel hedef, kişinin hastalığa sebebiyet veren etkenle karşılaştığında vücudunun etkeni tanıyarak hızlı bir şekilde bağışıklık oluşturması ve bu sırada hastalığa yakalanmamasıdır. Temel olarak (i) canlı zayıflatılmış aşılar ve (ii) inaktive aşılar olarak iki gruba ayrılabilen aşıların kendi içlerinde de birçok alt grubu bulunmaktadır .
Aşılanmanın kamu sağlığı açısından faydaları konusunda halkın bilinçlendirilmesi adına tüm dünyada olduğu gibi Türkiye'de de küçük yaşlardan itibaren hem çocuklar hem de yetişkinlere yönelik kamu sağlığı ve aşı eğitimleri verilmektedir. Kitle iletişim araçlarının da kamu sağlığı politikası aracı olarak kullanıldığı dikkate alındığında şu ana kadarki anlatımlarımızın aşina geldiği tahmin edilebilir. Bununla birlikte, son zamanlarda tüm dünyanın topyekün mücadele içerisine girdiği COVID-19 salgınına karşı ivedilikle aşı üretilmesi yönündeki yüksek beklenti, bir aşının keşfinden üretimine geçen sürede ne gibi regülasyonlara ve kurallara tabi olduğuna yönelik aşina olmadığımız bir sahaya girişimizi tetiklemiştir.
Esas itibarı ile aşılar da ilaç ve diğer biyolojik ürünlerdekine benzer şekilde birçok lisans ve üretim prosedürüne tabidir. Bununla birlikte aşıların etkinlik düzeyinin ilaçlarla karşılaştırıldığında çok daha yüksek olması aşılar için daha farklı regülasyonların söz konusu olmasına sebebiyet vermektedir.
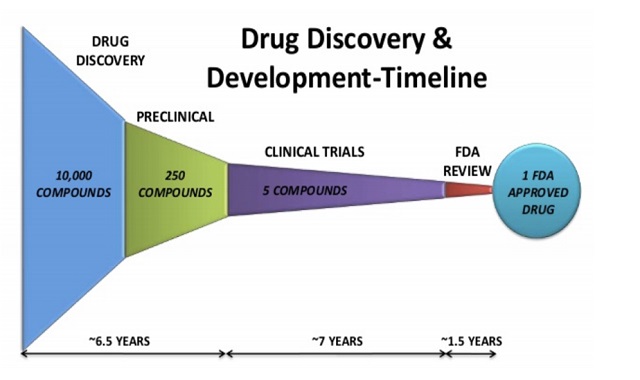
Bir aşının geliştirilmesi uzun yılları alabilir . Zira laboratuvar koşullarındaki bilimsel araştırmalarla başlayan süreç, belirli etik kurallar ve prosedürler ile yürütülen klinik çalışmalarla devam eder. Aşının etkinliğinin kanıtlanması sonrası üretim aşamasına geçilebilir. Bununla birlikte, üretim ve ithalatın tabi olduğu lisanslama süreçleri de söz konusu olabilir.
Örneğin, Türkiye'de ithal edilen
aşılar T.C. Sağlık Bakanlığı
Türkiye İlaç ve Tıbbi Cihaz Kurumunun
("TİTCK") yürüttüğü
ruhsatlandırma sürecine tabidir.
Tüm araştırmaların ve ruhsatlandırma
süreçlerinin tamamlanmasını müteakip
aşının olası yan etkilerini takip edilebilmesi
için "aşı sonrası istenmeyen
etkiler"e yönelik önceden belirlenmiş
prosedürler izlenmektedir.
En nihayetinde bir aşının kamuya uygulanabilmesi bakımından en önemli ölçüt olan "aşı etkinliği" aşının topluma uygulanmasıyla toplumda mücadele edilen belirli bir bulaşıcı hastalığın yüzde kaç azalacağını hesaplanmasıyla bulunabilmektedir. Genel olarak topluma önerilecek aşıların %90 gibi yüksek oranlarda etkinlik sağlaması beklenmektedir.
Kısacası bir aşının keşfinden, üretimine ve topluma uygulanmaya başlamasına geçen süreci düzenleyen bir dizi kural bulunmaktadır. Her ne kadar bu kuralların ulusal düzeyde ülkeden ülkeye belirli oranda farklılık arz edebileceği söylenebilecekse de uluslararası kabul gören belirli düzenlemelerin tüm dünyada esas alınarak uygulandığı görülmektedir.
Türkiye'de kullanılan tüm aşılar Dünya Sağlık Örgütü, Avrupa İlaç Kurumu (European Medicines Agency "EMA") ve ABD Gıda ve İlaç Kurumu'nun (The U.S. Food and Drug Adminisration "FDA") kurallarına uygun olarak ruhsatlandırılmış aşılardır . Bu kapsamda üretim süreçleri bakımından Dünya Sağlık Örgütü tarafından önerilen ve onaylanan İyi Üretim Uygulamaları (Good Manufacturing Practices) tüm dünyada kabul edilen kurallar getirirken aşı geliştirme çalışmalarına ilişkin temel prensipler bakımından ise FDA usul ve kurallarının dünyada genel olarak takip edildiğini görüyoruz. COVID-19 ile mücadelede tartışılan aşı ne zaman geliştirilecek ve toplumlar aşıya ne zaman kavuşabilecek gibi sorulara yönelik değerlendirmelere regülasyon bakımından ışık tutabileceği düşüncesiyle bu kuralların genel olarak ne gibi düzenlemeler olduğunu aşağıda sizin için derledik:
Aşı güvenliğine ilişkin öncü kurallar getirdiği kabul edilen FDA'nın özellikle lisanslama/ruhsatlandırma öncesi aşamalara ilişkin düzenlemeleri hemen hemen tüm dünyada kabul edilmekte ve ulusal düzenlemelere temel alınmaktadır. FDA'nın lisanslama öncesi kuralları (prelicensure) esas itibarı ile bir aşının ABD'de uygulanmasında önce geçmesi gereken test aşamalarını ifade etmektedir. Bu kapsamda bir aşı FDA tarafından onaylanmadan önce klinik araştırmaların ve tüm bilimsel çalışmaların FDA tarafından yetkilendirilmiş bilim adamları ve doktorlar tarafından en ince ayrıntısına kadar incelenmesi ve aşının güvenliği ile etkililiğinin yetkili heyet tarafından teyit edilmesi gerekmektedir. İlaveten, aşının lisanslama sonrası üretileceği tesislerin Dünya Sağlık Örgütü'nün İyi Üretim Uygulamaları'na uygunluk teşkil edip etmediği de FDA tarafından denetlenmektedir.

FDA kurallarına göre aşı geliştirme çalışmaları ilk olarak laboratuvar koşullarında yapılan bilimsel araştırmalar ve testlerle başlamaktadır. Laboratuvar koşullarında yapılan testler sonucu bir aşının bağışıklık yaratma potansiyeli olduğu görülürse hayvanlar üzerinde aşının test edilmesi aşamasına geçilmektedir. Hayvanlar üzerinde yapılan testler olumlu sonuç verir ve aşının insan üzerindeki bir test bakımından güvenli bir şekilde kullanılabileceğine ilişkin veriler alınabilirse gönüllü katılımcılar üzerinde aşının denenmesine ilişkin klinik test aşamasına geçilmektedir.
Gönüllü katılımcılar üzerinde yapılan klinik testler genel olarak üç aşamadan oluşmaktadır. İlk aşama (Phase I) 20 ila 100 arası sağlıklı katılımcı üzerinde yapılmakta ve "aşının güvenliği"ne odaklanmaktadır. İlk aşamanın önemli bir çıktısı hangi doz aralığının uygulanmasının güvenli olduğu sonucudur. Bu aşamada doz aralıklarına göre yan etki artışları da gözlemlenmektedir. Aşının etkili olup olmadığı ilk aşamanın aslî konusu değildir. Bununla birlikte, mümkün olduğu ölçüde aşının etkili olup olmadığı da gözlemlenmektedir.
İlk aşamada testleri sonlandırmayı gerektirecek ölçüde ve ciddiyette bir yan etkinin gözlemlenmemesi sonucunda ikinci aşamaya (Phase II) geçilmektedir. İkinci aşama birkaç yüz gönüllü katılımcının üzerinde aşının denenmesinden ibarettir. Bu aşamada genel olarak kısa dönemli yan etkiler gözlemlenmekte olup, aşı dozundaki değişime göre bağışıklık tepkisinin nasıl etkilendiği değerlendirilmektedir.
Klinik testlerin üçüncü aşaması (Phase III) ise "aşının etkililiği"ne yoğunlaşmaktadır. İkinci aşamadaki denek sayısından fazla olmak şartıyla genelde birkaç bin katılımcı üzerinde aşı denenmektedir. Bu aşamada farklı denek grupları oluşturulmakta ve aşının güvenliği denek gruplarının ortak özelliklerindeki farklılaşmaya göre test edilmektedir. Aşı etkinliği bakımından ise en önemli veri "placebo" ilaç olarak adlandırılan aslında deney konusu aşı olmayan bir ilacın belirli denek gruplara verilmesi ve deney konusu aşının verildiği grubun verdiği bağışıklık tepkisi ile placebo etkisinde olan grubun verdiği bağışıklık tepkisinin test edilmesi sonucu elde edilmektedir.
Klinik testlerin tüm aşamaları FDA'nın belirlediği plan doğrultusunda gerçekleştirilmekte ve FDA tarafından yetkilendirilen bilim heyetinin yakın gözetiminde yürütülmektedir. Bu kapsamda sıkı ve sürekli bir raporlama trafiği söz konusu olmaktadır.
Klinik testler ve bilimsel araştırmaların tüm sonuçları FDA'nın yetkili bilim heyeti tarafından değerlendirildikten sonra aşının lisanslanıp lisanslanamayacağına karar verilmektedir. Bu değerlendirmedeki temel ölçüt şudur; testleri tamamlanan aşının gözlemlenen faydaları, aşının önerileceği insan grubu üzerinde doğacak potansiyel sağlık risklerini bertaraf edebilecek düzeyde midir? Bu soruya yalnızca "evet, net fayda potansiyel risklerin üzerindedir" cevabı verilebildiği takdirde FDA tarafından bir aşıya lisans verilmekte ve aşının uygulanmasına izin verilmektedir.
Bununla birlikte, aşının uygulanmasına geçildikten sonra da aşının sonuçları yakın takibe alınmaktadır. Lisanslama sonrası (postlicensure) takip çalışmalarının iki temel sebebi bulunmaktadır: (i) Klinik deneyler, sık rastlanmayan yan etkilerin test edilebilmesi için yeterli büyüklükte bir denek grubunu içermeyebilir, (ii) klinik deneyler farklı denek grupları belirlenerek sürdürülse dahi bazı yan etkilerin söz konusu olabileceği denek gruplarını içermeme ihtimali bulunaktadır. Hamilelik, yaş grubu, kronik hastalıklar klinik testlerde dikkate alınan gruplar olsa da deneylerin her bir ihtimali ve farklılığı göz önüne alması mümkün değildir.
Lisanslama sonrası takip çalışmaları kapsamında klinik testlerde gözlemlenmeyen bir yan etkinin söz konusu olması halinde aşının etkililiği ve güvenliğine ilişkin lisanslamaya konu değerlendirme tekrar yapılmaktadır.
Lisanslama çalışmalarının bir ayağı da FDA tarafından üretim tesislerine ve faaliyetlerine yönelik yapılan incelemelerdir. Aşı üretimi "lot" denilen küçük gruplar halinde yapılmaktadır. Üretici, her bir lotun eşit derecede güvenli, saf ve etkili olmasını temin etmekle yükümlüdür. ABD'de üretilen aşı lotları FDA tarafından dolaşıma sokulmadığı müddetçe ticari dağıtıma konu edilememektedir.
Üretime ilişkin FDA tarafından yapılan değerlendirmeler daha önce de ifade ettiğimiz üzere Dünya Sağlık Örgütü'nün İyi Üretim Uygulamaları'na uyumluluk doğrultusunda yapılmaktadır.
İyi Üretim Uygulamaları (Good Manufacturing Practices "GMP" veya güncel hali için "cGMP") aşıların belirli kalite standartlarına uygun olarak üretilmesini ve aşılara ilişkin denetimde bu kalite standartlarının uygulanmasını temin etmeye yönelik kurulmuş bir sistemdir. Dünya Sağlık Örgütü tarafından belirlenen uygulamalar bütünü esas itibarı ile yalnızca aşı üretimi için değil, ilaç sektöründeki test aşamaları tamamlanmaksızın piyasaya sürülemeyen tüm ilaç ürünlerinin üretimi için getirilmiştir.
GMP, üretimde kullanılan ekipmanlardan ürün kullanımına ilişkin eğitime, etiketlemeden üretimde görev alacak olan personelin kişisel hijyenine üretim aşamasının tüm safhalarına yönelik kurallar öngörmektedir. GMP, ulusal otoriteler için doğrudan bağlayıcılık teşkil etmemekle birlikte Dünya Sağlık Örgütü'nün GMP'ye ilişkin yayınladığı ayrıntılı rehberler ulusal otoritelerin kendi iç denetim kurallarını nasıl dizayn etmeleri gerektiğine ışık tutmaktadır.
GMP, klinik testler veya bilimsel araştırmalarla önüne geçilmeyecek kalite kontrol hatalarını bertaraf etmeye yönelik tasarlanmıştır. Bu nedenle GMP'nin amacı bir aşının güvenli veya etkili olup olmadığına yönelik tıbbi açıdan denetleme mekanizmaları yaratmaktan ziyade şirketlerin üretim faaliyetlerini lisanslanan üründen beklenen faydayı sağlamaya uygun olarak şekillendirmesini temin etmeyi hedeflemektedir. Bu anlamda GMP, laboratuvar koşullarında test edilen ve lisanslanan ilaçların üretim bandında aynı kalitede ve güvenilirlikte çoğaltılmasını sağlamaktadır.
Oldukça ayrıntılı kurallar içeren GMP rehberleri Dünya Sağlık Örgütü'nün resmi internet sitesinde yayınlanmakta ve sürekli olarak güncellenmektedir. Bu rehberlere rağmen açıkta kalan konulara ilişkin olarak Dünya Sağlık Örgütü'nün ülke temsilcisiyle veya bölge ofisiyle öncelikli olarak iletişime geçilmesi, halen sorun çözülemiyorsa Cenevre'de bulunan merkeze başvurulması tavsiye edilmektedir.
Originally published 7 May, 2020
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
